Efficiently Inferring the Demographic History of Many Population with Allel Count Data pdf epub mobi txt 电子书 下载 2026
- 分子生物学
- Demographic History
- Population Genetics
- Allele Frequency
- Statistical Inference
- Computational Biology
- Genomics
- Evolutionary Biology
- Phylogeography
- Data Analysis
- Machine Learning

具体描述
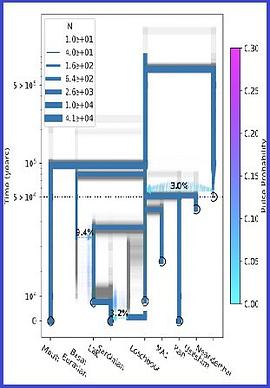
The sample frequency spectrum (SFS), or histogram of allele counts, is an important summary statistic in evolutionary biology, and is often used to infer the history of population size changes, migrations, and other demographic events affecting a set of populations. The expected multipopulation SFS under a given demographic model can be efficiently computed when the populations in the model are related by a tree, scaling to hundreds of populations. Admixture, back-migration, and introgression are common natural processes that violate the assumption of a tree-like population history, however, and until now the expected SFS could be computed for only a handful of populations when the demographic history is not a tree. In this article, we present a new method for efficiently computing the expected SFS and linear functionals of it, for demographies described by general directed acyclic graphs. This method can scale to more populations than previously possible for complex demographic histories including admixture. We apply our method to an 8-population SFS to estimate the timing and strength of a proposed "basal Eurasian" admixture event in human history. We implement and release our method in a new open-source software package momi2.
作者简介
目录信息
读后感
评分
评分
评分
评分
用户评价
这本书的价值,很大程度上来源于它对“数据类型”的深刻理解和利用。等位基因计数数据(Allele Count Data)相比于全基因组测序数据,在某些方面具有信息冗余度更高、存储和处理负担更轻的特点,尤其在涉及大规模群体比较时。这本书的精妙之处或许就在于,它找到了一种方法,能够从这些相对“粗糙”但获取成本低廉的数据中,最大限度地榨取出关于历史事件的信号。这涉及到一个核心的平衡艺术:如何在信息量和计算可行性之间找到最佳的甜蜜点。我对书中关于模型选择和假设检验的部分非常感兴趣。例如,在推断何时发生了种群分裂,或者种群规模发生了多大变化时,不同的统计模型可能会给出不同的解释。这本书是否提出了一套标准化的流程来评估这些模型的相对优劣,并指导读者根据数据本身的特性来选择最合适的推断工具?这种方法论上的成熟度,决定了这本书能否真正成为该领域的基石性著作,而非昙花一现的技巧集合。
评分我个人认为,这本书的出现恰逢其时,它瞄准了当前群体遗传学研究中的一个核心瓶颈:如何将“数据量大”的优势转化为“推断速度快”的优势。传统的基于马尔可夫链蒙特卡洛(MCMC)的方法在处理超大规模数据集时,往往需要数周甚至数月的时间进行收敛,这极大地限制了研究的迭代速度和探索性。因此,任何声称能“高效推断”的方法,必然涉及对计算复杂度的精妙控制,可能借鉴了近似推断(Approximate Inference)的思想,或者采用了新的优化算法,例如利用矩阵分解或特殊的概率分布来加速计算。我期待书中能详细阐述这些加速机制背后的数学原理,尤其是当群体数量(即“Many Populations”)显著增加时,算法的扩展性(Scalability)是如何得到保证的。这种对计算效率的执着追求,使得这本书的读者群体不再局限于理论生物学家,它也将吸引大量的生物信息学工程师和从事大规模数据分析的科研人员。
评分从阅读体验上来说,这本书的叙事逻辑组织得相当出色。它并没有一开始就抛出极其复杂的数学公式,而是循序渐进地引导读者进入核心思想。初学者可能会被其宏大的主题吓到,但一旦进入正文,就会发现作者精心设计的章节结构,使得复杂的概念逐步被拆解和阐明。比如,在介绍如何将原始的等位基因计数数据转化为可用于推断的统计量时,作者一定花费了大量篇幅来解释数据预处理的必要性与合理性,这部分内容对于确保下游分析的有效性至关重要。我特别关注到,作者如何巧妙地平衡理论深度与实际操作性之间的关系。理论推导固然重要,但若不能转化为实际可运行的算法,便只是空中楼阁。因此,我推测书中对算法实现、计算效率优化以及软件工具的引用或探讨,占据了相当比重,这使得这本书既有学术价值,又有很强的工程实践指导意义。它不只是一本理论手册,更像是一本“实战指南”,教会我们如何在高通量数据时代,真正实现“高效”这一目标。
评分这本关于群体遗传学和人口历史推断的著作,着实让人眼前一亮。它的核心吸引力在于其对海量等位基因计数数据的高效处理能力。在如今基因组学研究中,数据量呈爆炸式增长,如何从中提炼出可靠的人口历史信息,是摆在研究者面前的巨大挑战。这本书似乎提供了一种优雅且计算上可行的方法论。我特别欣赏它在理论建模上的严谨性,它不仅仅停留在描述性的统计分析,而是深入到了基于概率模型的推断框架。这种深度使得其方法不仅适用于教科书式的简单场景,更能应对真实世界中复杂、高维度的群体结构。想象一下,面对来自全球数千个独立群体的等位基因计数矩阵,传统方法可能因计算复杂度而望而却步,而这本书所倡导的“高效推断”理念,无疑为我们打开了一扇新的大门。这种高效性并非以牺牲准确性为代价,书中必然包含对模型假设的深入探讨和误差分析,确保推断结果的稳健性。对于任何从事复杂群体结构分析的研究人员来说,这本书无疑是一份宝贵的资源,它预示着我们能够以前所未有的速度和规模,描绘出人类乃至其他物种的宏大迁徙与分化图景。
评分这本书对于理解人类或物种传播的历史进程,提供了工具上的飞跃。当我们不再被计算资源所束缚时,我们就可以设计出更精细、包含更多群体和更多历史事件的复杂模型。我设想这本书的内容会启发我们去探索一些过去难以触及的问题,比如:在某一特定历史时期,是否存在一个未被充分采样的“中间群体”?或者,不同地理区域的群体分离时间是否存在系统性的偏差?这种对细微历史信号的敏感性,正是高效推断带来的附加价值。它不仅仅是更快,而是能提供更精细、更少偏差的视图。书中对模型不确定性的处理方式也值得称道——一个真正严谨的科学著作,不会只给出一个点估计的结果,而会量化推断结果的置信区间。这种对不确定性的坦诚处理,是科学推断成熟的标志,也确保了基于这些分析得出的历史结论,具有更高的可靠性和可信度。
评分 评分 评分 评分 评分相关图书

















本站所有内容均为互联网搜索引擎提供的公开搜索信息,本站不存储任何数据与内容,任何内容与数据均与本站无关,如有需要请联系相关搜索引擎包括但不限于百度,google,bing,sogou 等
© 2026 book.quotespace.org All Rights Reserved. 小美书屋 版权所有